Your new post is loading...
COVID-19 Symptoms Swayed By Genetics More info from WIRED Magazine Some people experience Covid-19 as nothing more than a mild cold, and others exhibit no symptoms at all. Then there are the thousands who sicken and, often, die. Scientists are working hard to understand the underlying reasons for such huge discrepancies in symptoms and outcomes. No one knows the answer yet. One theory: It is locked deep in our genetic makeup. We know that age and underlying health conditions, such as hypertension, play a large role in determining how people fare once they’ve contracted Covid-19. But these alone don’t explain the wide diversity of symptoms. Studying the genetics of the virus and people who are more susceptible to SARS-CoV-2 could not only help identify and protect those more at risk but also help speed treatment and drug development. “What is it that makes some people very sick and other people hardly sick at all? There are two major possibilities,” says Kári Stefánsson, head of deCODE Genetics, an Icelandic subsidiary of Amgen Inc. that has conducted some of the most extensive studies of the virus to date. One is the genetic sequence of the virus itself: that some strains make people sicker than others, he says. The other: the unique genetics of each person who catches the disease. Some people’s genes may simply make them more vulnerable to severe illness, while others’ genetics may confer resistance. It is generally accepted that our genes do play a role in how we respond to viral infections. On the extreme end, one mutation of the gene CCR5, for example, makes those who carry it resistant to human immunodeficiency virus, or HIV. Certain genetic variants, especially in genes that influence the immune system, seem to predispose people to a host of other infectious diseases. One 2017 study looked at 23 common infections including chickenpox, shingles and cold sores and found genes that seemed to be associated with many of them. Stefánsson and other scientists suspect human genetic variations may play a similar role in people who suffer from Covid-19. There are some early indications of this with the novel coronavirus. The receptor it uses to penetrate host cells, called ACE2, can be present in varying numbers in different people based on their genetics and on environmental factors, such as what medicines they take.
Researchers at Washington University School of Medicine in St. Louis have used induced pluripotent stem cells produced from the skin of a patient with a rare, genetic form of insulin-dependent diabetes, transformed the stem cells into insulin-producing cells, used the CRISPR gene-editing tool to correct a defect that caused the diabetes, and implanted the cells into mice to reverse diabetes in the animals. Using induced pluripotent stem cells produced from the skin of a patient with a rare, genetic form of insulin-dependent diabetes called Wolfram syndrome, researchers transformed the human stem cells into insulin-producing cells and used the gene-editing tool CRISPR-Cas9 to correct a genetic defect that had caused the syndrome. They then implanted the cells into lab mice and cured the unrelenting diabetes in those mice. The findings, from researchers at Washington University School of Medicine in St. Louis, suggest the CRISPR-Cas9 technique may hold promise as a treatment for diabetes, particularly the forms caused by a single gene mutation, and it also may be useful one day in some patients with the more common forms of diabetes, such as type 1 and type 2. The study is published online April 22 in the journal Science Translational Medicine. Patients with Wolfram syndrome develop diabetes during childhood or adolescence and quickly require insulin-replacement therapy, requiring insulin injections multiple times each day. Most go on to develop problems with vision and balance, as well as other issues, and in many patients, the syndrome contributes to an early death. "This is the first time CRISPR has been used to fix a patient's diabetes-causing genetic defect and successfully reverse diabetes," said co-senior investigator Jeffrey R. Millman, PhD, an assistant professor of medicine and of biomedical engineering at Washington University. "For this study, we used cells from a patient with Wolfram syndrome because, conceptually, we knew it would be easier to correct a defect caused by a single gene. But we see this as a stepping stone toward applying gene therapy to a broader population of patients with diabetes."
Original announcement In a population of animals or plants, genetic diversity can decline much more quickly than species diversity in response to various stress factors: disease, changes to habitat or climate, and so on. Yet not much is known about fish genetic diversity around the world. Help on that front is now on the way from an international team of scientists from French universities and ETH Zurich. They have produced the first global distribution map for genetic diversity among freshwater and marine fish. Furthermore, they identified the environmental factors that are instrumental in determining the distribution of genetic diversity. Their study was recently published in the journal Nature Communications. Genetic diversity is unevenly distributed To begin their study, the researchers analyzed a database that contained the data of over 50,000 DNA sequences representing 3,815 species of marine fish and 1,611 species of freshwater fish. From this sequence data, the scientists estimated the average genetic diversity in sections of bodies of water, each section measuring 200 square kilometers. Their analysis revealed that genetic diversity is unevenly distributed throughout marine and freshwater fish. The greatest genetic diversity was found among marine fish in the western Pacific Ocean, the northern Indian Ocean and the Caribbean. Among freshwater fish, genetic diversity was greatest in South America, but comparatively low in Europe. In addition, the researchers determined that temperature is a key factor influencing genetic diversity among marine fish: as the temperature rises, so does diversity. By contrast, the key determinants of genetic diversity in freshwater fish were the complexity of their habitat structure and how their habitats have changed over time.
A new gene therapy appears to serve as a functional cure for the most common type of hemophilia, early clinical trial results indicate. Patients who received the one-time intravenous therapy continue to have a more than 90 percent decrease in bleeding events two to three years after their initial treatment, researchers reported Jan. 1, 2020 in the New England Journal of Medicine. The therapy fixes a broken gene in liver cells that causes production of flawed factor VIII, a protein that plays a key role in blood clotting. People with this genetic mutation have hemophilia A, the most common type of this bleeding disorder. Hemophilia A accounts for 8 out of 10 cases of hemophilia, researchers said. Hemophilia A patients must inject themselves with factor VIII every other day to prevent bleeding, said lead researcher John Pasi, a professor at Barts and The London School of Medicine and Dentistry in England. "There's been a massive reduction in bleeding in the patients, and none of them any longer need to regularly treat themselves with factor VIII to prevent bleeding," Pasi said of participants in the phase 1/phase 2 clinical trial. "That huge treatment burden of having to give yourself an intravenous injection every other day has gone away." The therapy uses a virus to carry the DNA sequence for a functional factor VIII gene into liver cells, he said. "It infects liver cells and transfers into those cells the factor VIII gene," Pasi said. "Liver cells make factor VIII and then secrete it, and it passes into the circulation." Seven initial participants in the study have a 96 percent decrease in bleeding events three years out, researchers report. Another six who joined later had a 92 percent decrease in bleeding by the end of year two. "At three years, the patients who were treated at a higher dose were expressing functional levels of factor VIII -- somewhat lower than they were at their peak, but they're still at really good levels that are hugely effective in protecting the patients against bleeding," Pasi said.
The majority of our human genome transcribes into noncoding RNAs with unknown structures and functions. Obtaining functional clues for noncoding RNAs requires accurate base-pairing or secondary-structure prediction. However, the performance of such predictions by current folding-based algorithms has been stagnated for more than a decade. Scientists now propose the use of deep contextual learning for base-pair prediction including those noncanonical and non-nested (pseudoknot) base pairs stabilized by tertiary interactions. Since only <<250 nonredundant, high-resolution RNA structures are available for model training, they utilized transfer learning from a model initially trained with a recent high-quality bpRNA dataset of >>10,000 nonredundant RNAs made available through comparative analysis. The resulting method achieves large, statistically significant improvement in predicting all base pairs, noncanonical and non-nested base pairs in particular. The proposed method (SPOT-RNA), with a freely available server and standalone software, should be useful for improving RNA structure modeling, sequence alignment, and functional annotations.
Collecting DNA samples for human genetic studies can be an expensive, lengthy process that has often made it difficult to include diverse populations in studies of medical and health data. University of Michigan researchers and their colleagues believe they have found a way to harness the power of social media and its ubiquitous presence to recruit a large, diverse participant pool they hope will help provide quick, reliable data for genetic studies. Their study appears in the June 13, 2019 issue of The American Journal of Human Genetics. “The ability to study very large groups of individuals is a key challenge in human genetics, which is using very rare genetic changes—each present in very few individuals—to understand human biology and health and provide leads for design of new medicines,” said senior author Gonçalo Abecasis, a professor at U-M’s School of Public Health. Katharine Brieger, a doctoral student in public health and first author of the report, said that for studies to be relevant to a broader population, they need to include samples of a wide range of racial, ethnic and socioeconomic backgrounds. “Historically, genetic studies were largely made up of people who lived near university medical centers, inadvertently excluding people who lived in more remote areas or who didn’t have the time and money to travel,” she said. “Allowing remote participation with Genes for Good allows many of these people to participate in research for the first time.” Researchers invited people to participate in the Genes for Good study through Facebook starting in January 2015. Requirements included that participants live in the United States, have a Facebook account and be at least 18. Most of the recruitment was done organically, with people finding the Genes for Good application through family and friends. As of March 2019, about 117,000 people tried the app, 80,000 people had engaged with the study, 32,000 kits had been sent and 27,000 DNA samples collected. Genotypes for the first 20,232 participants were analyzed.
Surviving fragments of genetic material preserved in sediments allow metagenomics researchers to see the full diversity of past life — even microbes. The moment an organism dies, its body begins to decompose. Its cells rupture, and their contents spill into the environment. What scientists have realized over the past two decades is that even though the physical structure of a body disappears, its DNA can last for centuries. Eske Willerslev, an evolutionary geneticist at the University of Copenhagen, has found this DNA in the least likely places, including the soil underneath glaciers, in caves and more. His work has helped rewrite the natural histories of many locations around the globe by reconstructing ecosystems as much as 450,000 years old, and maybe older. He claims the idea came to him as a graduate student. “It was the autumn, and I saw leaves falling from the trees, and I saw a dog take a crap on the street,” he recalled, all of which set him to wondering: “‘What happens to that DNA? Could it somehow be preserved in the sediments or in the soil where it was put?’”
Via Complexity Digest
Viruses, the most abundant biological entities on earth, are a scourge on humanity, causing both chronic infections and global pandemics that can kill millions. Yet, the true extent of viruses that infect humans remains completely unknown. Some newly discovered viruses are recognized because of the sudden appearance of a new disease, such as SARS in 2003, or even HIV/AIDS in the early 1980s. New techniques, however, now enable scientists to identify viruses by directly studying RNA or DNA sequences in genetic material associated with humans, enabling detection of whole populations of viruses—termed the virome—including those that may not cause acutely recognizable disease. However, identifying novel types of viruses is difficult as their genetic sequences may have little in common with already known viral genomes that are available in reference databases. Researchers in the Perelman School of Medicine at the University of Pennsylvania, have now identified a previously unknown viral family, which turns out to be the second-most common DNA virus in human lung and mouth specimens, where it is associated with severe critical illness and gum disease, respectively. The team published their findings today in Cell Host Microbe. Senior authors Frederic D. Bushman, Ph.D., chair of the department of Microbiology, and Ronald G. Collman, MD, a professor of Pulmonary, Allergy and Critical Care, lead the team that discovered this new virus, which they dubbed Redondoviridae. "New sequencing techniques have helped us uncover a world of new viruses," said Bushman. "However, the majority of the sequence data we have so far remains unclassified, leaving us much work to do in order to better understand the human virome and how these new species may be associated with illness." The team analyzed samples from human lungs to sequence RNA and DNA in free-floating viral particles. Using this wide-ranging survey of viral sequences as a starting point to compare samples with known viral sequences in public databases, the team, including first authors Arwa A. Abbas and Louis J. Taylor, both graduate students working with Bushman and Collman, identified short sequences that were similar to a type of virus found in domesticated pig stool. From there, they identified whole genomes in the sample, which they recognized as a completely new human virus.
Via Emilio Mordini, Juan Lama
The genealogist’s dream of testing old, spit-laced artifacts is coming true—but raising questions about who controls dead people’s DNA. Last fall, Gilad Japhet, the founder of a DNA-testing company, got up at an industry conference to talk about his grandmother Rosa’s love letters. Japhet’s company, MyHeritage, sells cheek swabs to people interested in their family history. It now has 2.5 million people in its DNA database, making it the third largest behind 23andMe and AncestryDNA. But Japhet wasn’t satisfied with only testing the living; he wanted to test the dead. Which brings us to the love letters—or really, the envelopes they came in. The envelopes were sealed by his grandmother, and the stamps on them presumably licked by her. “Maybe our ancestors did not realize it,” Japhet said, a smile growing on his face, “when they were licking those stamps and the envelope flaps, they were sealing their precious DNA for you forever.” Then he made the big announcement: MyHeritage would soon begin offering DNA testing on old stamps and envelopes. He didn’t stop there. If you can test the letters of your grandmother, why not those of historical figures? Japhet is a prodigious collector of autographs, and he revealed that he possessed handwritten letters from Albert Einstein and Winston Churchill. In an intriguing if provocative PR move, he promised that “their DNA is coming to MyHeritage very, very soon.” In the past year, genealogists have been abuzz about the possibility of getting DNA out of old stamps and envelopes. In addition to MyHeritage, a British company called Living DNA began informally offering the service for $400 to $600 last year, and a small Australian start-up called Totheletter DNA, which specializes in DNA from envelopes and stamps, launched a similarly priced service in July. MyHeritage says its own service should debut later this year. (A spokesperson declined to comment on when Einstein and Churchill’s DNA profiles will be uploaded to the company’s site.)
The great white shark is one of the most recognized marine creatures on Earth, generating widespread public fascination and media attention, including spawning one of the most successful movies in Hollywood history. This shark possesses notable characteristics, including its massive size (up to 20 feet and 7,000 pounds) and diving to nearly 4,000 foot depths. Great whites are also a big conservation concern given their relatively low numbers in the world’s oceans. Longevity clues are tucked away in the great white shark's genome. Certain adaptations identified in the fish’s DNA linked to wound healing, cancer protection, and a long life. In a major scientific step to understand the biology of this iconic apex predator and sharks in general, the entire genome of the white shark has now been decoded in detail. A team led by scientists from Nova Southeastern University’s (NSU) Save Our Seas Foundation Shark Research Center and Guy Harvey Research Institute (GHRI), Cornell University College of Veterinary Medicine, and Monterey Bay Aquarium, completed the white shark genome and compared it to genomes from a variety of other vertebrates, including the giant whale shark and humans. The findings are reported in the ‘Latest Articles’ section of the journal Proceedings of the National Academy of Sciences, USA.
After sequencing one kākāpō's genome, the Department of Conservation's (DOC) kākāpō scientist, Dr Andrew Digby, came up with the idea to sequence every single kākāpō's genome. After a long, complicated process - which included collecting blood from every kākāpō, and a crowdfunding campaign to raise money, which was coordinated by the Genetic Rescue Foundation, a non-profit that aims to preserve biodiversity through genetic techniques - it has now been completed. In the time that lapsed, several kākāpō died, but many more were hatched; What began with a target of 125 genomes has become 172, including all 147 living birds (and several dead ones). It marks the second time globally an entire species has had its genome mapped - The first, the Spix's macaw, is believed to be extinct in the wild - and has the potential to shake up what we thought we knew about kākāpō. "We've basically got the set of all genes, of all the individuals, in the entire species," Digby said. "It's a massive resource... It's kind of limitless, in a way, what we can find out." Few bird species worldwide have attracted as much publicity and research interest as the kākāpō, but it remains frustratingly enigmatic. Some of the outstanding questions are complicated. Why, for example, are some birds more prone to particular diseases, like the dreaded "crusty bum", than others? But other questions are simple, and speak to how little we know about the ancient parrot. Chief among those questions: How long does a kākāpō live for? The answer is likely a very long time, but until now, there has been no way to know for sure. Around 32 living birds were discovered in the wild as adults - known as "founders" - meaning their age is unknown. "We know they're at least 35 or 40 years old, but are they 50? Or 100?" Digby said. "Are they about to die next year? We've got no idea." Some of those kākāpo may well be 100 years old. Perhaps the most famous kākāpo, Richard Henry, was found near Milford Sound in 1975 and died in 2010. He was the only mainland kākāpo to breed, providing precious genetic diversity to a population otherwise entirely drawn from Stewart Island.
In April, a citizen scientist named Barbara Rae-Venter used a little-known genealogy website called GEDMatch to help investigators find a man they’d been looking for for nearly 40 years: The Golden State Killer. In the months since, law enforcement agencies across the country have flocked to the technique, arresting a flurry of more than 20 people tied to some of the most notorious cold cases of the last five decades. Far from being a forensic anomaly, genetic genealogy is quickly on its way to becoming a routine police procedure. At least one company has begun offering a full-service genetic genealogy shop to law enforcement clients. And Rae-Venter’s skills are in such high demand that she’s started teaching her secrets to some of the biggest police forces in the US, including the Federal Bureau of Investigation. Identifying individuals from their distant genetic relatives, a technique called long-range familial searching, is a potent alternative to the types of DNA searches commonly available to cops. Those are typically limited to forensic databases, which can only identify close kin—a sibling, parent, or child—and are highly regulated. No court order is required to mine GEDMatch’s open source trove of potential leads, which, unlike forensic databases, contains genetic bits of code that can be tied to health data and other personally identifiable information. Currently, there aren’t any laws that regulate how law enforcement employs long-range familial searching, which hobbyists and do-gooders have turned to for years to find the biological families of adoptees. But some legal experts argue its use in criminal cases raises grave privacy concerns. They expect to see a legal challenge at some point, though probably not in the next year. In the meantime, GEDMatch is becoming even more powerful, as it grows by nearly a thousand new uploads every day. And with hundreds more cases currently in the hands of full-time family-tree builders, one thing’s for sure: In 2019, genealogy is going to send a lot more people to jail.
The genomes of two long-lived giant tortoises, including Lonesome George, reveal candidate genes and pathways associated with their development, gigantism and longevity. Lonesome George's species may have died with him in 2012, but he and other giant tortoises of the Galapagos are still providing genetic clues to individual longevity through a new study by researchers at Yale University, the University of Oviedo in Spain, the Galapagos Conservancy, and the Galapagos National Park Service. Genetic analysis of DNA from Lonesome George and samples from other giant tortoises of the Galapagos—which can live more than 100 years in captivity—found they possessed a number of gene variants linked to DNA repair, immune response, and cancer suppression not possessed by shorter-lived vertebrates. The findings were reported Dec. 3 in the journal Nature Ecology & Evolution In 2010, Caccone began sequencing the whole genome of Lonesome George, the last of the species Chelonoidis abingdonii, to study evolution of the tortoise population on the Galapagos. Carlos Lopez-Otin at the University of Oviedo in Spain analyzed this data and other species of tortoises to look for gene variants associated with longevity. "We had previously described nine hallmarks of aging, and after studying 500 genes on the basis of this classification, we found interesting variants potentially affecting six of those hallmarks in giant tortoises, opening new lines for aging research," Lopez-Otin said.
|
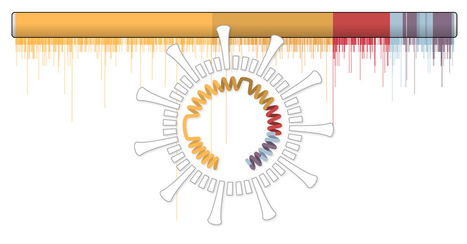
The virus has mutated. But that doesn’t mean it’s getting deadlier. At this point in the pandemic, coronavirus genomes with 10 or fewer mutations are common, and only a small number have over 20 mutations — which is still less than a tenth of a percent of the genome. Over time, viruses can evolve into new strains — in other words, viral lineages that are significantly different from each other. Since January, researchers have sequenced many thousands of SARS-CoV-2 genomes and tracked all the mutations that have arisen. So far, they haven’t found compelling evidence that the mutations have had a significant change in how the virus affects us.
Comprehensive genomic characterization of tumours became a major goal of cancer researchers as soon as the first human genome had been sequenced in 2001. Since then, advances in sequencing technology and analytical tools have allowed this research field to flourish. In six papers1–6 in this issue of Nature, the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium presents the most comprehensive and ambitious meta-analysis of cancer genomes so far. Unlike previous efforts that focused largely on protein-coding regions of the cancer genome, PCAWG analyses whole genomes. Each article scrutinizes an important aspect of cancer genetics — together, their findings will be key to understanding the full genetic complexity of cancer. Before discussing the impact of these analyses, it is crucial to highlight the massive amount of data and the complex organizational framework that underpin the PCAWG endeavor. The project involved an interdisciplinary group of scientists from 4 continents, with 744 affiliations between them, who had to overcome major technical, legal and ethical challenges to carry out distributed analyses while protecting patient data. Researchers were divided into 16 working groups, each focused on distinct facets of cancer genomics — assessing the recurrence of mutations, for instance, or inferring tumor evolution. Altogether, the consortium performed integrative analyses of 38 tumor types. The group sequenced 2,658 whole-cancer genomes (Fig. 1), alongside matched samples of non-cancerous cells from the same individuals. These data were complemented by 1,188 transcriptomes — the sequences and abundances of RNA transcripts in a tumor.
UC San Diego researchers studied nearly 900 vertebrate species and found that bats have unusual gut microbiomes that more closely resemble those of birds than other mammals, raising questions about how evolutionary pressures change the gut microbiome. Diet and host phylogeny drive the taxonomic and functional contents of the gut microbiome in mammals, yet it is unknown whether these patterns hold across all vertebrate lineages. Now, researchers assessed gut microbiomes from ∼900 vertebrate species, including 315 mammals and 491 birds, assessing contributions of diet, phylogeny, and physiology to structuring gut microbiomes. In most nonflying mammals, strong correlations exist between microbial community similarity, host diet, and host phylogenetic distance up to the host order level. In birds, by contrast, gut microbiomes are only very weakly correlated to diet or host phylogeny. Furthermore, while most microbes resident in mammalian guts are present in only a restricted taxonomic range of hosts, most microbes recovered from birds show little evidence of host specificity. Notably, among the mammals, bats host especially bird-like gut microbiomes, with little evidence for correlation to host diet or phylogeny. This suggests that host-gut microbiome phylosymbiosis depends on factors convergently absent in birds and bats, potentially associated with physiological adaptations to flight. These findings expose major variations in the behavior of these important symbioses in endothermic vertebrates and may signal fundamental evolutionary shifts in the cost/benefit framework of the gut microbiome.
Just two years ago, GEDmatch was still an obscure genealogy website, known only to a million or so hobbyist DNA sleuths looking to fill in their family trees. The site was free, public, and run by two guys with a knack for writing algorithms that helped relatives find each other. All in all, it was a pretty controversy-free place. That all changed in April of 2018, when news broke that police had used GEDmatch to identify a suspect in the 40-year-old Golden State Killer case. As the site emerged as a crime-fighting tool, some users and privacy experts began to worry about how people’s genetic data might ensnare them in criminal investigations, when all they wanted was to learn about their family history. The transition has been rocky for GEDmatch. One drama after another has engulfed the website: Police searches have grown increasingly invasive; the site’s owners tried to react with changes to its terms of service that ended up backfiring; and white-hat hackers pointed out glaring security flaws. But starting Monday, that’s all someone else’s problem. Early December 2019, GEDmatch announced it was being taken over by a new owner, the forensic genomics firm Verogen. The San Diego-based company spun out of sequencing giant Illumina two years ago, specializing in next-generation DNA testing services catered to law enforcement. With the acquisition of GEDmatch, Verogen may also start offering genealogy searches like the ones that have so far identified suspects in as many as 70 cases. “Never before have we as a society had the opportunity to serve as a molecular eyewitness, enabling law enforcement to solve violent crimes efficiently and with certainty,” Verogen CEO Brett Williams said in a statement announcing the deal. The terms of the agreement were not disclosed. Reactions so far, have been mixed. “I suspect this will be the last straw for all the genealogists who don’t want to share with law enforcement,” Debbie Kennett, a genealogist and honorary research associate at University College London, told WIRED. On Monday GEDmatch updated its terms of service to reflect the new ownership, but it did not alert users via email. Kennett found out from a Facebook group discussion. When she tried to log into GEDmatch, she discovered she was locked out until she accepted the new terms. Additional options included deciding later and permanently deleting all her data from the GEDmatch servers. According to a Verogen spokesperson, whatever settings users had earlier selected for their GEDmatch profiles—opting in or out of police searches—will remain under the new terms. GEDmatch itself has not always stuck to its word on such matters. Earlier this month, the site’s users discovered their privacy settings weren’t ironclad, when reports surfaced that a Florida detective had obtained a warrant to search the site’s full database, including individuals who had opted out of cooperating with law enforcement. A few days prior, a team of genetic security researchers revealed a flaw in GEDmatch’s relative-matching algorithm that would allow a hacker to scrape more than 90 percent of users’ DNA data. Verogen’s Williams says GEDmatch has already addressed these security issues, and that his company will continue to monitor other possible vulnerabilities.
Researchers in Sweden and the U.S. have devised a new method for studying individual cells in human tissue, which could lead to even earlier detection of diseases such as cancer and ALS. The method offers a 1,400-fold increase in spatial resolution. Published in the scientific journal, Nature Methods, the study was performed by researchers from KTH Royal Institute of Technology and the Broad Institute of MIT and Harvard. The technology builds on transcriptomics, or the mapping of the cell's molecular fingerprints. RNA-sequencing provides a tool for studying different functions within tissues but the molecular profile only reveals so much. The cell's position in the tissue also has several stories to tell, including how different diseases occur. Methods for studying active genes in individual cells from tissue have evolved dramatically. At the forefront of this research are various microfluidic systems where researchers have been able to isolate individual cells and then carry out so-called massive parallel RNA-sequencing. This method has enabled the profiling of hundreds of thousands of cells from tissue, but provides no information about the cell's origin in the tissue. At the same time, other spatial and molecular methods that capture detailed information about the position of cells in frozen or solid tissue have been developed. Yet such methods still demand knowledge of which genes—or biomarkers—researchers want to investigate, which in turn requires relatively large and expensive instruments. In addition, it is usually only possible to study hundreds of genes at one time, out of thousands of active ones.
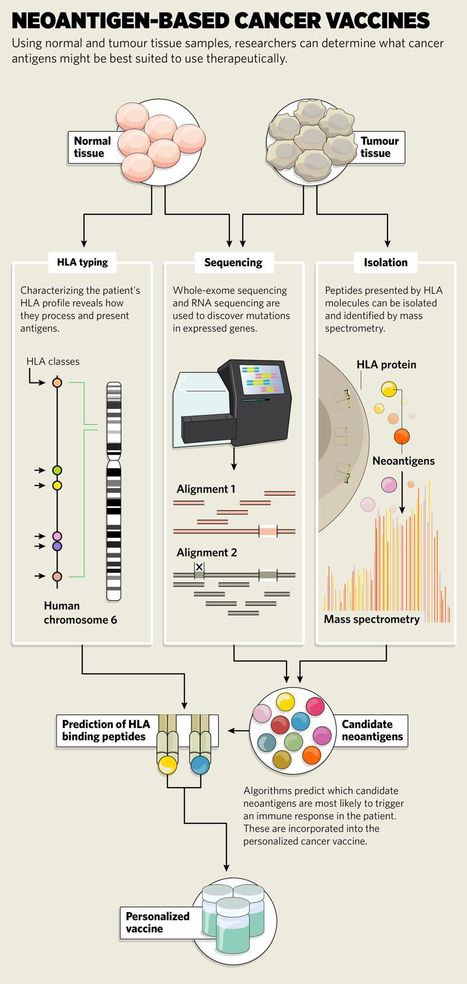
Next-generation sequencing technologies are helping researchers to find mutations unique to an individual’s cancer as well as the genetic signatures that predict their immune response. Can they use these clues to develop long-lasting and effective anticancer vaccines? Cancer vaccines that make use of these mutations represent a promising therapeutic strategy. “By stimulating immune responses against tumors through vaccines, we have an opportunity to generate long-term anti-tumor immunity,” says Benjamin Vincent, an immune-oncologist in the Division of Hematology/Oncology at the University of North Carolina at Chapel Hill. Not only do vaccines limit the damage to healthy cells, they can also prevent cancer recurrence by training the immune system to respond to those cells in the future. Vincent is using immunogenomic approaches in his research to understand tumor biology and develop clinically relevant biomarkers and new cancer therapies. Cancer-associated mutations frequently lead to the production of neoantigens: unique protein fragments that the immune system recognizes as foreign. Consequently, the total number of mutations in cancer cells (the tumor mutational burden, TMB) is emerging as a biomarker for predicting response to immunotherapy across all cancer types. “TMB is now used as a surrogate for the number of predicted neoantigens,” he says. Tumors with a high TMB are more likely to respond to immune checkpoint inhibitor drugs, although this has not been found in all cancer types1. “Even if neoantigens are present in the tumor, there can be multiple reasons for immunotherapy failure,” says Vincent. These reasons include the patient’s T cells not recognizing the neoantigens, or their neoantigen-recognizing T cells are somehow suppressed or unable to reach the tumor site. Checkpoint inhibitors, which interfere with cancer cells’ mechanism for evading the immune response, are likened to ‘releasing the brakes’ of the immune system. But these drugs fail to eradicate the tumor in the majority of cases. In these instances, it could be more effective to ‘hit the accelerator’ by priming the immune system to kill cancer cells directly, using a vaccination strategy that targets their mutant proteins. “The hope is that, by both expanding the repertoire of neoantigen-recognizing T cells with a vaccine and removing the brakes that tumor cells put on the immune system with checkpoint inhibitors, we can obtain a synergistic effect,” says Vincent.
Via Krishan Maggon
A research team consisting of scientists from Kyoto University, Tokyo University of Science, National Institute for Physiological Sciences, and Tokyo Institute of Technology, report in the Journal of Virology the Medusavirus, a unique giant virus that gives pause to current theory on viral evolution. The name Medusavirus was given for the effect this virus has on its host, Acanthamoeba castellanii. Once infected, the amoeba forms cysts, a phenomenon called encystment. This is a typical response to environments hostile to survival, and leaves the amoeba with a hard, protective covering. Perhaps it was not a coincidence then that Medusavirus was found in the hot springs in northern Japan, the first giant virus to have been isolated from a heated environment. Along with the location of its discovery, Medusavirus holds a number of distinguishing features compared with other giant viruses. Its DNA codes for all five types of histones, the key proteins that help compact DNA within the nucleus. In fact, no other known virus has all five types. Further, Medusavirus encoded neither RNA polymerase nor DNA topoimerase II, whereas all other giant viruses encode at least one. These features could explain why the replication of Medusavirus DNA begins and completes in the host nucleus to eventually fill the amoeba nucleus with viral DNA, which again is unlike other giant viruses. Moreover, the morphology of the capsid surface was unique, in that it was covered with an extraordinary number of spherical-headed spikes. In addition, the amoeba genome encoded several capsid surface proteins. The existence of histone genes in Medusavirus and capsid protein genes in amoeba suggest lateral gene transfer going both directions -- host-to-virus and virus-to-host. Overall, the findings suggest that Medusavirus offers a new model for host-virus co-evolution and that the Medusavirus is a new family of large DNA viruses.
Via Juan Lama
Genetic testing for pets is expanding. Hundreds of thousands of dogs have now been genetically screened, as Petunia was, and companies are beginning to offer tests for cats. But the science is lagging. Most of these tests are based on small, underpowered studies. Neither their accuracy nor their ability to predict health outcomes has been validated. Most vets don’t know enough about the limitations of the studies, or about genetics in general, to be able to advise worried owners. Pet genetics must be reined in. If not, some companies will continue to profit by selling potentially misleading and often inaccurate information; pets and their owners will suffer needlessly; and opportunities to improve pet health and even to leverage studies in dogs and cats to benefit human health might be lost. Ultimately, people will become more distrustful of science and medicine.
Blue-blooded and armored with 10 spindly legs, horseshoe crabs have perhaps always seemed a bit out of place. First thought to be closely related to crabs, lobsters and other crustaceans, in 1881 evolutionary biologist E. Ray Lankester placed them solidly in a group more similar to spiders and scorpions. Horseshoe crabs have since been thought to be ancestors of the arachnids, but molecular sequence data have always been sparse enough to cast doubt. University of Wisconsin-Madison evolutionary biologists Jesús Ballesteros and Prashant Sharma hope, then, that their recent study published in the journal Systematic Biology helps firmly plant ancient horseshoe crabs within the arachnid family tree. By analyzing troves of genetic data and considering a vast number of possible ways to examine it, the scientists now have a high degree of confidence that horseshoe crabs do indeed belong within the arachnids. "By showing that horseshoe crabs are part of the arachnid radiation, instead of a lineage closely related to but independent of arachnids, all previous hypotheses on the evolution of arachnids need to be revised," says Ballesteros, a postdoctoral researcher in Sharma's lab. "It's a major shift in our understanding of arthropod evolution." Arthropods are often considered the most successful animals on the planet since they occupy land, water and sky and include more than a million species. This grouping includes insects, crustaceans and arachnids. Horseshoe crabs have been challenging to classify within the arthropods because analysis of the animals' genome has repeatedly shown them to be related to arachnids like spiders, scorpions, mites, ticks and lesser-known creatures such as vinegaroons. Yet, "scientists assumed it was an error, that there was a problem with the data," says Ballesteros.
The Mexican tetra is a creature that spend most of its live in dark caves after its ancestors swam from surface waters into their caves around 1.5 million years ago. Like naked mole rats—another species that lives underground, in perennial darkness—they have no eyes. But whereas key genes controlling eye development in naked mole rats have mutations that inactivate them, there are no such inactivating mutations in the genes of the Mexican cave fish. But mutations aren't the only way to change gene activity, and new research suggests a different explanation for the fish's lack of eyes. Epigenetic regulation is a means of controlling gene activity that does not alter the DNA sequence of the genes themselves. Genes undergoing epigenetic regulation can still make normal proteins, but the amount of the protein they make is modulated. One method of epigenetic regulation is the addition of methyl groups to the DNA that controls the activity of specific genes. It is efficient, since it can be used to alter a whole bunch of genes at once. And it seems to explain the Mexican cave fish eye degeneration that has occurred over the past million years—a proverbial blink of an eye in evolutionary terms. These cave fish have elevated levels of an enzyme that adds methyl groups to DNA compared to surface fish. This enzyme, called a DNA methylase, is expressed in the developing eye, where it decorates genes important in eye development with methyl groups. This reduces their expression to about half of the levels seen in surface fish. A number of the methylated genes are known to be lost or defective in human eye disorders like colorblindness, night blindness, and even complete blindness. One of these genes (Pax6) controls the expression of a whole host of other eye genes, amplifying the effect. There's evidence that altered methylation is central to the changes in these fish. A drug called 5-azacytidine (5-AzaC) is a potent inhibitor of DNA methylation (it's used by people with myelodysplastic syndrome, a rare disorder in which blood cells do not mature properly). Injections of the drug into cave fish embryo eyes could partially rescue their development, confirming that their loss is due to changes in methylation rather than mutations or gene loss. While this sounds like an evolutionary change that isn't reliant on mutations, that may not be the case. The genetic mechanism responsible for the upregulation of the methylating enzyme in cave fish is not yet known. It's likely that some changes in DNA sequences were involved there. Nature Ecology and Evolution, 2018. DOI: 10.1038/s41559-018-0569-4 (About DOIs).
Adolf Hitler's deputy flew to Scotland in 1941 and was imprisoned for the rest of his life. But was the man in Spandau really Rudolf Hess? Now a DNA test has revealed the truth The Deputy Führer of the Third Reich Rudolf Hess was captured after a controversial flight to Scotland in 1941. Hess was sentenced to life imprisonment during the Nuremberg War Crimes Trials. He was detained in Berlin’s Spandau Prison under the official security designation ‘Spandau #7.’ Early doubts arose about the true identity of prisoner ‘Spandau #7.’ This evolved to a frequently espoused conspiracy theory that prisoner ‘Spandau #7’ was an imposter and not Rudolf Hess. After Hess’s reputed 1987 suicide, the family grave became a Neo-Nazi pilgrimage site. In 2011, the grave was abandoned and the family remains cremated. Here scientists now report the forensic DNA analysis of the only known extant DNA sample from prisoner ‘Spandau #7’ and a match to the Hess male family line, thereby refuting the Doppelgänger Theory. The prisoner in Spandau #7 was indeed Rudolf Hess.
Natural hair color within European populations is a complex genetic trait. Previous work has established that MC1R variants are the principal genetic cause of red hair color, but with variable penetrance. Here, a group of geneticists have now extensively mapped the genes responsible for human hair color in the Caucasian British ancestry, participants in UK Biobank. MC1R only explains 73% of the SNP heritability for red hair in the UK Biobank, and in fact most individuals with two MC1R variants have blonde or light brown hair. The scientists identified several other genes contributing to red hair, the combined effect of which accounts for ~90% of the SNP heritability. Blonde hair is associated with over 200 genetic variants and the researchers find a continuum from black through dark and light brown to blonde and account for 73% of the SNP heritability of blonde hair. Many of the associated genes are involved in hair growth or texture, emphasizing the cellular connections between keratinocytes and melanocytes in the determination of hair color.
|